Jannis Wilts und Jan Ole Schlenger
12.10.2025 bis zum 24.10.2025
Im Rahmen eines Stipendiums der Auricher Wissenschaftstage durften wir ein zweiwöchiges Herbstpraktikum am Institute of Structural Biology in Bonn absolvieren. Forschende des Instituts beschäftigen sich vorrangig mit der Analyse biomedizinisch relevanter Strukturen. Hierbei handelt es sich in der Regel um eine Expression zellulär gebildeter Proteine. Dabei werden multiple Verfahren aus der Biochemie und Molekularbiologie genutzt, um die Struktur und Funktionsweise ausgewählter Proteine zu ermitteln.
Diese Art der Forschung findet besonders Anwendung in der Pharmazie und Medizin, da sie die Grundlage der Herstellung von Medikamenten bildet. Aufgrund der Möglichkeit, Zellen genetisch zu verändern, kann hierbei besonders auf Mutationen bereits bekannter Proteine eingegangen werden.
Während des Praktikums durften wir Einblicke in die Forschungsprojekte verschiedener Mitglieder des Forschungsteams von Prof. Dr. Matthias Geyer erhalten. Der Großteil der Forschung widmete sich der Strukturanalyse menschlicher Inflammasome.
Unser erster Tag begann nach einer kurzen Vorstellung direkt mit einem „Labmeeting“. Hierbei handelt es sich um ein rein organisatorisches online-Meeting aller Angestellten des Labors (aller Forschungsgruppen), welches wöchentlich stattfindet. Anschließend erhielten wir eine Führung durch alle von der Forschungsgruppe genutzten Labore im Biomedizinischen Zentrum 1.

Nach der Führung fand ein gruppeninternes Meeting statt, in dem ein Mitglied der Gruppe Geyer den Fortschritt seines aktuellen Projektes beleuchtete. Danach wurde über mögliche Verbesserungen und andere Herangehensweisen diskutiert. Thema des Projekts war erneut ein Teilbereich der Inflammasom-Forschung. Dabei ging es vor allem um die Aktivierung dieses Proteins. Der Einfachheit halber stellen wir die verschiedenen Abläufe und Methoden, die wir in den folgenden zwei Wochen kennenlernten, in einer chronologischen Reihenfolge dar, wie sie auch in einem beispielhaften Projekt bestehen würde.
Normalerweise beginnt ein Projekt in der Strukturbiologie mit einer PCR, bei der wir DNA-Plasmide mithilfe eigens am Computer erstellter Primer genetisch verändert haben. Während der PCR werden ausgewählte DNA-Sequenzen in drei sich wiederholenden Schritten vermehrt. Der erste Schritt ist die Denaturation. Bei 98 °C Celsius werden die DNA-Doppelstränge in Einzelstränge aufgetrennt, sodass die hinzugegebenen Primer an den richtigen Sequenzen binden können. Das Binden der Primer (Annealing) findet bei 72 °C statt. Darauf folgt die Verlängerung (Elongation) ebenfalls bei 72 °C. Beigefügte DNA-Polymerasemoleküle synthetisieren den jeweils komplementären DNA-Strang. Hierzu wird ein Thermocycler verwendet, der die Änderungen in der Temperatur zwischen den einzelnen Schritten ermöglicht.
Aufgrund einzelner veränderter Basen in den erstellten Primern können bei diesem Schritt Mutationen in die DNA eingebaut werden. Die PCR stellt den Anfang der Expression eines Proteins dar und ist somit die Grundlage fast jeder strukturbiologischen Forschung.

Als Teil eines anderen Projektes haben wir die auf die PCR folgende Transformation kennengelernt. Es werden die in der PCR hergestellten Plasmide in kompetente Zellen eingesetzt, damit die Zellen das Zielprotein exprimieren. In unserem Fall handelte es sich hierbei um E. Coli-Bakterien, häufig werden aber auch Insektenzellen verwendet. Die Bakterien werden durch einen Hitzeschock für die Aufnahme der Plasmide empfänglich gemacht und nach einer einstündigen Inkubationszeit bei 37°C auf Agaroseplatten ausgestrichen. Am nächsten Tag werden von dieser dann einzelne Bakterienkolonien „gepickt“ und in flüssigem Nährmedium suspendiert.
Anschließend widmeten wir uns dem in der Regel auf eine Transformation folgenden Schritt, der Aufreinigung des Zielproteins. Da zur weiteren Verwendung nur das Protein, welches durch die eingesetzten Plasmide exprimiert wurde, benötigt wird, muss dieses aus den Zellen gelöst (lysiert) werden. Zunächst werden dazu die Zellen mithilfe von Ultraschall in einem speziellen Gerät namens „Sonicator“ aufgebrochen. Nach der Zugabe von Protease-Inhibitoren und DNAse wird dann die Lösung zentrifugiert. Die aufgebrochenen Zellen setzen sich als ein Pellet ab, während alle zelleigenen Proteine in einer darüberliegenden Flüssigkeit gelöst sind.
Die gefilterte Lösung wird anschließend nach dem Zielprotein aufgereinigt. Im Zuge einer Chromatographie an der “Äkta Start“ wird die Proteinlösung durch eine mit einem speziellen Zucker gefüllte Säule geleitet. Äkta Start ist ein Gerät zur automatischen Reinigung von Proteinen, bei dem eine Proteinlösung durch eine spezielle Trennsäule geleitet wird. Aufgrund einer TAG genannten Änderung der Struktur des Zielproteins während der Expression bindet dieses an dem in der Säule enthaltenen Stoff, wobei die anderen Proteine durch diese hindurch gespült werden. Mithilfe einer Pufferlösung, die das gebundene Protein wieder aus der Säule löst, wird das Zielprotein schließlich in kleine Proberöhrchen gespült. Durch die Trennung in diese Fraktionen wird gewährleistet, dass das Protein nicht durch den Puffer verdünnt wird.

Als nächstes stand im Labor die Analyse der Proteine an. Zunächst haben wir mithilfe einer weiteren Chromatographie an der “Äkta Pure“ das bereits aufgereinigte Zielprotein in einer Probenlösung nachgewiesen. Äkta Pure ist ein flexibles Chromatographie-System zur Reinigung und Trennung von Proteinen mithilfe verschiedener Säulentypen. Dabei werden die verschiedenen Bestandteile der Lösung aufgrund ihrer Größe und chemischen Eigenschaften voneinander getrennt. Die Leitfähigkeit und UV-Absorption der austretenden Lösung werden gemessen, um das gewünschte Protein nachzuweisen.
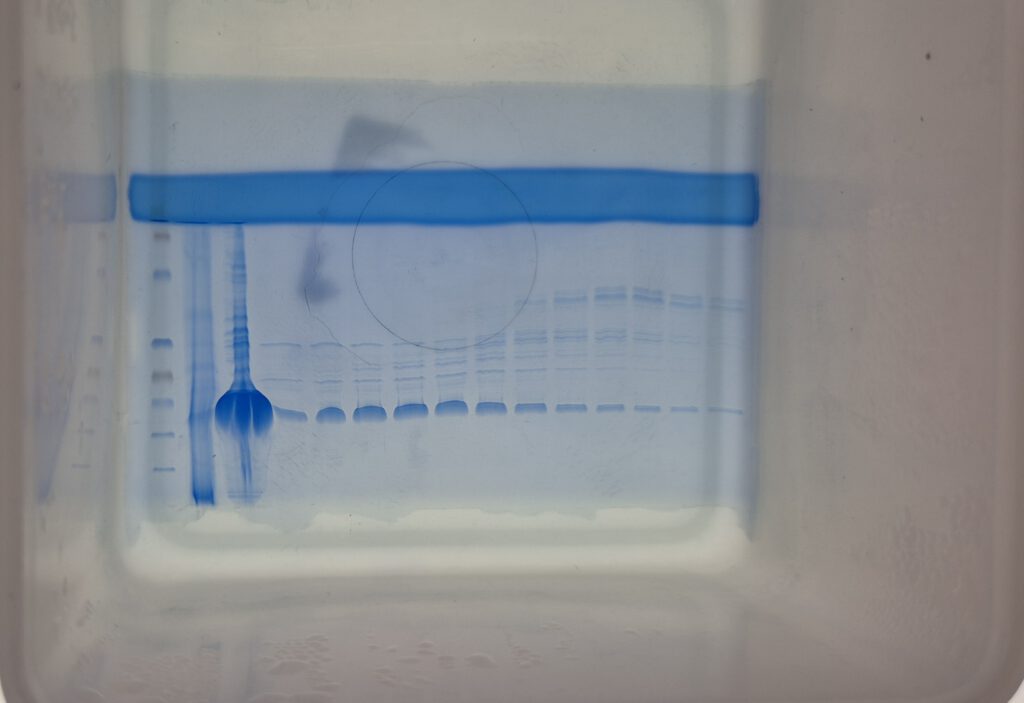
Im Anschluss folgte eine Gelelektrophorese mit einem SDS-Gel. Hierbei wandern Proteine unter elektrischer Spannung in einer gewissen Zeit unterschiedlich weit durch ein Gel. Nach dem Färben konnten die entstandenen Banden ausgewertet und die Reinheit der Proben überprüft werden.
Aus den Proben mit dem höchsten Reinheitsgrad wird dann eine sogenannte Stock-Lösung erstellt, deren Konzentration zunächst bestimmt werden muss. Dies geschieht, indem ca. 2µl der Stock-Lösung auf ein Gerät (Nanodrop) getropft werden, welches den entstandenen Tropfen durchleuchtet und anhand der Lichtbrechung die Proteinkonzentration bestimmt.
Um die Konzentration zu erhöhen, wird die Lösung in ein Amicon- Röhrchen gegeben. Dieses enthält einen Filter, der für die Pufferlösung durchlässig ist, das Protein jedoch zurückhält. Durch Zentrifugation wird die Lösung durch den Filter gedrückt und es bleibt eine hoch konzentrierte Proteinlösung im oberen Teil des Amicons zurück.

Eine Methode, um die Struktur des Zielproteins zu bestimmen, ist die CRYO EM. Eine Proteinprobe wird im gefrorenen Zustand unter dem Rasterelektronenmikroskop betrachtet. Die Vorbereitung für den Transport in eine solche Einrichtung geschieht in der Regel durch eine “Plunge-Freezing“ genannte Methode, bei der eine Maschine sowohl mit flüssigem Stickstoff als auch mit flüssigem Ethan-Gas befüllt wird. Anschließend wird das einzufrierende Protein auf ein äußerst kleines
Carbongitter, auch Grit genannt, pipettiert, welches in die Maschine eingespannt ist. Dieses wird dann in das Behältnis mit flüssigem Ethan gestürzt und schlagartig auf ca. -160°C heruntergekühlt. Hierdurch wird verhindert, dass sich beim Einfrieren große Eiskristalle bilden, die ein deutliches Bild unter dem Mikroskop verhindern würden. Gelagert und transportiert werden die Grits in flüssigem Stickstoff bei ca. -180°C.
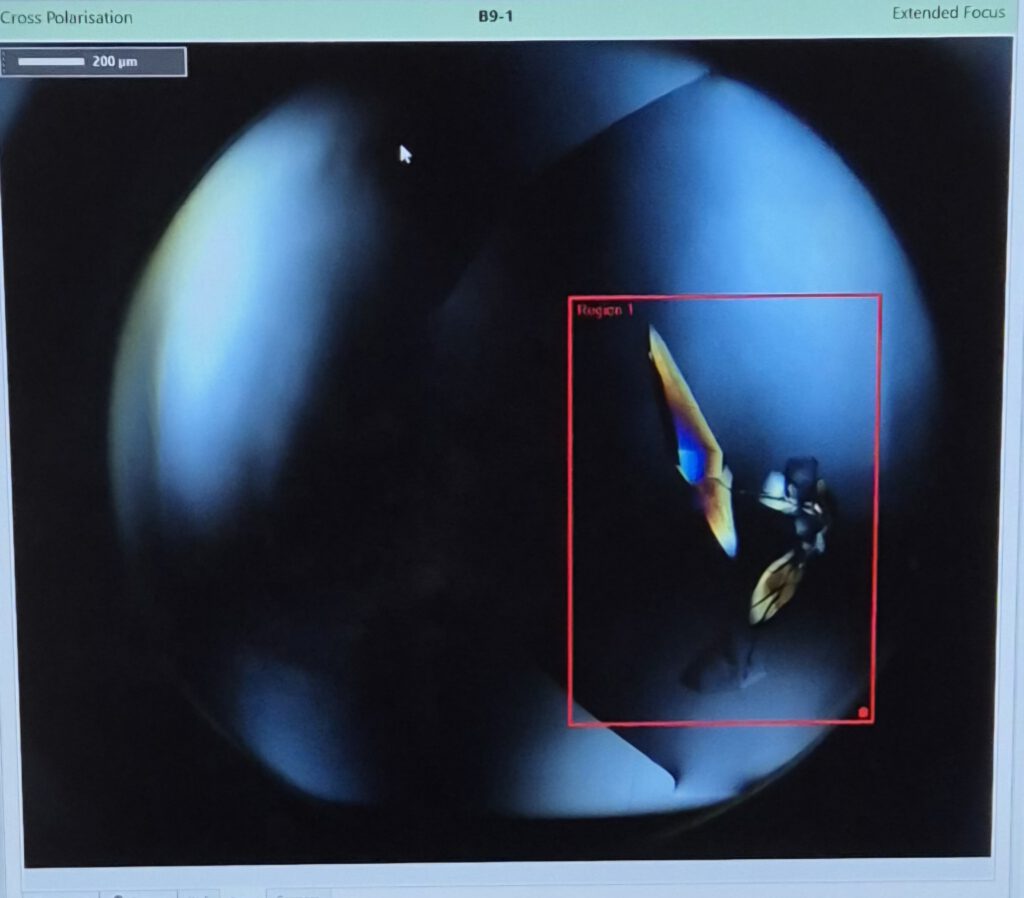
An einem anderen Tag stand der Bereich der Kristallographie im Mittelpunkt. Diese Methode ermöglicht es, die räumliche Struktur von Proteinen zu bestimmen und dadurch besser zu verstehen, wie sie aufgebaut sind und wie sie funktionieren. Um solche Strukturdaten zu gewinnen, müssen Proteine zunächst in Kristallform gebracht werden, ein Vorgang, bei dem sich die Moleküle regelmäßig anordnen und feste Strukturen bilden.
Im Labor wurde uns gezeigt, wie dieser Prozess technisch umgesetzt wird. Mithilfe einer Maschine namens “Gryphon“ konnten winzige Mengen Proteinlösung automatisiert in verschiedene Bedingungen pipettiert werden, um optimale Kristallisationsvoraussetzungen zu finden. Anschließend wurde an einer weiteren Maschine ein Bild der Kristallstrukturen aufgenommen, um sichtbar zu machen, wie die Kristalle geordnet sind und sie so für spätere Röntgenanalysen vorzubereiten.
Zum Abschluss durften wir selbst Kristalle „fischen“, also sie mit einer feinen Nadel vorsichtig aus der Lösung entnehmen und auf kleine Träger platzieren. Dabei konnten wir praktisch erleben, wie präzise und sorgfältig in der Kristallographie gearbeitet wird und welche Schritte notwendig sind, um die Struktur von Proteinen sichtbar zu machen.
Eine andere Möglichkeit, mehr über ein aufgereinigtes Protein herauszufinden, ist die “Surface Plasmon Resonance“ (SPR), eine Methode, die die Bindungsfähigkeiten von Proteinen analysiert. Hierbei kann mithilfe von Lichtreflektion gemessen werden, ob bestimmte Proteine an andere Proteine oder auch andere Stoffe binden. Da dieser Prozess relativ kompliziert ist, verbrachten wir den Dienstag mit der Vorbereitung der Messungen, parallel wurde uns der Ablauf genauer erläutert: Das Bindungsvermögen eines zu untersuchenden Proteins wird gemessen, indem ein besonderer Chip in ein Gerät geladen wird. Auf diesem Chip befindet sich eine feine Goldschicht, die auf einer Seite von einer reflektierenden Glasscheibe und auf der anderen Seite von einer Dextranzuckerschicht bedeckt ist. Das SPR-Gerät beschießt die verglaste Seite des Chips mit Licht und misst den Winkel, in dem das reflektierte Licht zurückgeworfen wird. Wird nun ein Protein auf dem Chip immobilisiert, indem es in einer Pufferlösung über die Dextranschicht gespült wird und an diese bindet, ändert sich aufgrund der erhöhten Teilchenmasse an dieser Stelle auch der Winkel, in dem das Licht reflektiert wird. Diese Verschiebung des Reflektionswinkels kann gemessen werden. Wird nun ein zweiter Stoff über den Chip gespült, kann festgestellt werden, ob dieser an das immobilisierte Protein bindet oder nicht. Ändert sich die Lichtreflektion erneut, spricht dies für eine Veränderung der Masse, was wiederum bedeutet, dass der neu hinzugegebene Stoff an das immobilisierte Protein gebunden ist. Die Stärke des gemessenen Ausschlags lässt darauf schließen, wie gut und schnell die beiden Stoffe aneinanderbinden.


Da bei unserem Versuch von Bedeutung war, dass das untersuchte Inflammasom NLRP 3 in einer ganz bestimmten Ausrichtung auf dem Chip immobilisiert wird, damit eine spezielle Bindestelle, auch Bindetasche genannt, „frei“ bleibt, verwendeten wir einen besonderen Chip, bei dem bereits ein Stoff namens Streptavidin an das Dextran gebunden war. Unser NLRP3-Protein war in der Expression so verändert worden, dass es nur für die Messung relevante Teile aufwies, zusätzlich jedoch auch den Stoff Biotin beinhaltete. Biotin bildet eine starke Bindung zu Streptavidin aus, sodass das Protein immer mit dieser Stelle an den Chip bindet und immobilisiert wird.
Neben der Arbeit mit Bakterien- und Insektenzellen haben wir uns auch mit menschlichen Zellen befasst. Hierfür ging es für uns ins S2-Labor (erhöhte Sicherheitsstufe), in dem abgesehen von menschlichen Zellen auch an Viren geforscht wird. Ziel des Projektes war es, herauszufinden, inwiefern entzündungshemmende Medikamente in ihrer Wirkung eingeschränkt sind, wenn ein bestimmtes Inflammasom (NLRP3) als Mutation vorliegt. Dafür wurden kleine Mengen von Zellen in Probefelder einer Platte hinein pippettiert und in unterschiedlichen Zeitabständen mit unterschiedlichen Medikamenten behandelt. Anschließend haben wir eine “Flow Cytometry“-Messung durchgeführt, während der die einzelnen Zellen einer Probe in einer Maschine mit Lasern beschossen werden. Anhand der Absorption und Reflexion des Lichts können Vorhandensein und Menge des vorher mit einem Farbstoff markierten Inflammasoms festgestellt werden.
Der Donnerstag begann für uns mit einem Besuch in der Zellkultur. Dort werden Zellstämme genetisch unveränderter Zellen versorgt und vermehrt. In unserem Fall handelte es sich hierbei um besonders geeignete Insektenzellen. Diese werden in den verschiedenen Schritten der Proteinexpression, die wir bereits an den vorherigen Tagen kennenlernen durften, benötigt, da beim Einsetzen von Erbgut in Zellen, um die Synthese eines Proteins hervorzurufen, immer vom genetischen Wildtyp ausgegangen werden muss, um Verfälschungen der Ergebnisse zu vermeiden. Die gesamte Einrichtung benötigt daher regelmäßig neue und unbehandelte Zellen. Um einheitliche Forschungsbedingungen und einen hohen Hygienestandard zu gewährleisten, findet die Pflege der Zellkulturen zentral in einem eigenen Labor statt. Nach einer gründlichen Einweisung bezüglich des sterilen Arbeitens, bekamen wir zunächst gezeigt, wie die Stammlösungen versorgt und geteilt werden. Da eine Kontamination an dieser Stelle äußerst ungünstig wäre, konnten wir bei diesem Schritt nur zusehen. Jedoch durften wir anschließend die Zelldichte in den einzelnen Stammlösungen aus Proben ermitteln und aus den verbliebenen Stammlösungen für unterschiedliche Projekte benötigte Lösungen mit unterschiedlichen Konzentrationen von Zellen herstellen.
Außerdem haben wir die Isolation von DNA-Plasmiden aus den Bakterien, denen wir am Vortag im Zuge der Transformation verändertes Erbgut eingesetzt haben, durchgeführt. Nach dem Aufschluss der Zellen mit einem Lysepuffer haben wir die Plasmid-DNA über mehrere Zentrifugations- und Waschschritte isoliert und anschließend in einer Lösung gesammelt. Diese DNA kann später für weitere molekularbiologische Analysen verwendet werden. In fast jedem der beschriebenen Schritte werden Pufferlösungen verwendet. Diese werden benötigt, um Proteine zu lösen und zu stabilisieren. Dabei müssen Salzgehalt, pH-Wert und Additive bei jedem Protein variiert werden. Wir durften während unserer Zeit in Bonn mehrere dieser Lösungen herstellen. Die genaue Konzentration hatten wir bereits am Vortag selbst berechnet, da sie für jede Verwendung individuell abgestimmt werden muss. Im Verlauf der ersten Woche folgten Fachgruppen übergreifende Meetings, in denen Mitglieder der Forschungsgruppe Geyer beispielsweise ihren Fortschritt während des Aufenthalts in einer Forschungseinrichtung in Australien vorstellten. Auch hierbei handelte es sich wieder um die Erforschung desselben Inflammasoms.
Zum Abschluss möchten wir uns herzlich bei allen bedanken, die uns während der beiden Praktikumswochen begleitet und unterstützt haben. Ein besonderer Dank gilt unseren Betreuerinnen und Betreuern, die sich viel Zeit genommen haben, uns die einzelnen Arbeitsschritte verständlich zu erklären und uns aktiv in die praktischen Tätigkeiten einzubeziehen sowie Prof. Dr. Matthias Geyer, der dieses Praktikum als Leiter des Instituts erst ermöglicht hat.
Ebenso möchten wir den Auricher Wissenschaftstagen danken, die uns diese spannende Möglichkeit eröffnet haben. Durch das Programm konnten wir Einblicke in verschiedene Forschungsbereiche gewinnen, von der biochemischen Laborarbeit über die Proteinaufreinigung bis hin zur Kristallographie. Insgesamt waren die beiden Wochen eine sehr wertvolle und lehrreiche Erfahrung, die uns einen realistischen Eindruck vom Alltag in der Forschung vermittelt und unser Interesse an naturwissenschaftlichen Themen weiter gestärkt hat.